
Równania reakcji połówkowych: problematyczne reakcje redoks ze związkami organicznymi
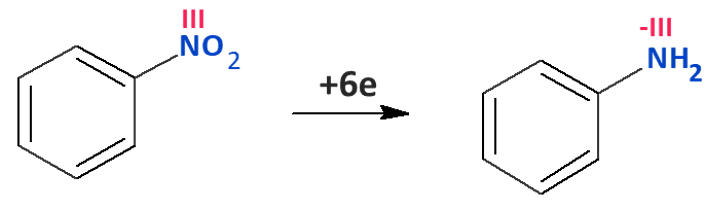
Zanim zaczniemy wspomnę tylko, że niektórzy nie potrafią zbilansować równania redukcji nitrobenzenu do aniliny, bo dochodzą do sprzeczności. Dlaczego dochodzą do sprzeczności? To sobie wyjaśnimy na samym końcu.
Choć w tym artykule skupimy się na związkach organicznych i pewnych problematycznych, albo nieoczywistych reakcjach redoks, to rzecz jasna podział na związki organiczne i nieorganiczne jest sztuczny i objawem wstecznictwa. Poruszone zagadnienia dotyczą tak samo związków organicznych, jak i nieorganicznych.
Pierwszą nieoczywistą reakcją redoks, którą sobie rozpatrzymy, jest nitrowanie arenów. Sumaryczne równanie reakcji nitrowania benzenu wygląda tak:
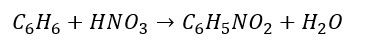
Ale choć często nie zdajemy sobie z tego sprawy, to ta reakcja jest reakcją redoks. A jak reakcja redoks, to i równania połówkowe… Jak zapisać równania reakcji utleniania i redukcji? Musimy wprowadzić grupę NO2 do cząsteczki benzenu jednocześnie usuwając atom wodoru. Jak zacząć zapisywanie równań połówkowych? Zacznijmy od wygenerowania indywiduum, które przyłączy się do cząsteczki benzenu w miejsce atomu wodoru:
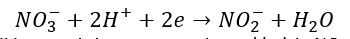
I mamy równanie redukcji i mamy już grupę atomów o składzie NO2. Następnie podstawiamy ja za 1 z atomów wodoru:
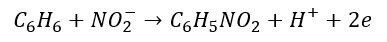
I mamy równanie utleniania! Po dodaniu reakcji połówkowych otrzymujemy:
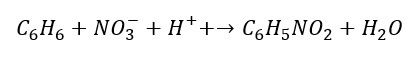
Czyli w zapisie cząsteczkowym:
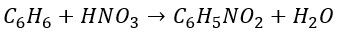
I teraz się zacznie: „Co ten Mickiewicz znowu wymyślił! Innych poucza, a sam nie wie, że w reakcji nitrowania benzenu bierze udział jon nitroniowy NO2+, a nie azotanowy(III)! Tak zgadzam się z tym, napisałem nieprawdę. I wiem o tym. A jednak ją napisałem.
Pójdźmy dalej. Jeśli napiszę równanie reakcji redukcji podczas utleniania alkoholi zakwaszonym roztworem dichromianu potasu:
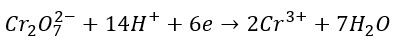
To napiszę tak samo prawdziwą, czyli nieprawdziwą informację o tej reakcji, jak w przypadku nitrowania. „Jak to! Przecież każdy powinien wiedzieć, że aniony dichromiannowe są silnymi utleniaczami i redukują się do kationów Cr3+!”
Tak to prawda, redukują się, a jednak napisałem nieprawdę. I to w obu przypadkach. Wynika to stąd, że to nie aniony dichromianowe(VI) działają utleniająca na alkohole, tylko aniony wodorochromianowe(VI), które powstają w układzie reakcyjnym. Po silnym zakwaszeniu wodnego roztworu dichromianu(VI) potasu ustala się równowaga:
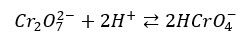
O tym, że taka równowaga ustala się w zakwaszonym roztworze dichromianu potasu świadczy kolor mieszaniny. Po dodaniu silnego kwasu do wodnego roztworu dichromianu potasu obserwujemy zmianę barwy z pomarańczowej na niemal czerwonopomarańczową. I to ten anion atakuje cząsteczkę alkoholu. Czyli właściwym utleniaczem nie jest anion dichromianowy!
Dlaczego zatem w równaniu połówkowym tego nie uwzględniamy? Bo równania połówkowe to fikcja. One nie odzwierciedlają w żaden sposób ścieżki procesu. Są tylko informacją o jego stechiometrii. O stanie początkowym i o stanie końcowym, a nie o mechanizmie reakcji. Tak więc tak samo jak w reakcji nitrowania benzenu nie bierze udziału anion azotanowy(III), tylko nitroniowy, to tak samo anion dichromianowy nie bierze udziału w reakcji utleniania alkoholi. I korzystając z tego samego prawa, wg którego zapisujemy równanie połówkowe z anionem dichromianowym, tak samo możemy zapisać równanie redukcji anionu azotanowego(V) do azotanowego(III) w przypadku nitrowania arenów.
Co więcej, to gdy wiemy, że równania połówkowe są fikcją, to w części przypadków równanie jednej reakcji połówkowej możemy zapisać na więcej niż jeden sposób. I jest to legitne. Ale o tym to za chwilę. Teraz wyjaśnijmy sobie raz na zawsze, że związki nitrowe nie są pochodnymi kwasu azotowego(V). Nie są. Gdyby były pochodnymi kwasu azotowego.(V), to byśmy je nazywali azotanami(V), a nie związkami nitrowymi. Błędne jest myślenie, że nitrobenzen jest pochodną kwasu azotowego(V), bo w reakcji jego otrzymywania użyto kwasu azotowego(V). To jest tylko metoda, którą go otrzymano. Ale związki nitrowe otrzymuje się także w innych reakcjach. Nitrowanie węglowodorów aromatycznych przebiega według mechanizmu substytucji elektrofilowej, stąd w reakcji potrzebny jest kwas azotowy(V), bo jest on źródłem kationu nitroniowego - właściwego elektrofila.
W innych przypadkach związki te można otrzymać albo według mechanizmu rodnikowego lub nukleofiwego. Nitrowanie alkanów prowadzi się w fazie parowej. Pary kwasu azotowego(V) i odpowiedniego alkanu reagują ze sobą, tworząc nitroalkany. W podniesionej temperaturze cząsteczki kwasu azotowego ulegają rozkładowi na rodniki:
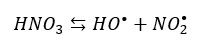
Nitrowanie alkanów przebiega więc według mechanizmu rodnikowego.
To jeszcze nie wszystko, ponieważ związki nitrowe możemy otrzymać w reakcji substytucji nukleofilowej. Klasycznej substytucji nukleofilowej. Chlorowcopochodne węglowodorów reagują z azotanami(III) i tworzą nitrozwiązki.
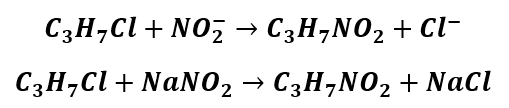
Tak, nitrowanie arenów jest reakcją redox. Ale wiele osób w pierwszej chwili odpowie, że to nie jest reakcja redox. To właśnie z tego powodu, że stopień utlenienia atomu azotu w grupie nitrowej wynosi III, związki litrowe można otrzymać w reakcji z azotanami(III). A czy reakcja chloroetanu z roztworem NaNO2 jest też reakcją redoks czy nie?
Rozpatrzmy jeszcze reakcję bromowania fenolu. W wodnym roztworze brom reaguje z fenolem tworząc 2,4,6-tribromofenol:
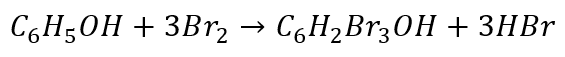
Jak zapisać równania połówkowe? Zacznijmy od redukcji…
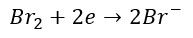
A co z równaniem utleniania? Możemy użyć części anionów bromkowych w tym równaniu, które wcześniej wygenerowaliśmy w równaniu redukcji.
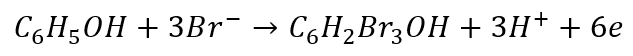
Po wymnożeniu równań połówkowych w celu zbilansowania elektronów i dodaniu otrzymujemy równanie sumaryczne:
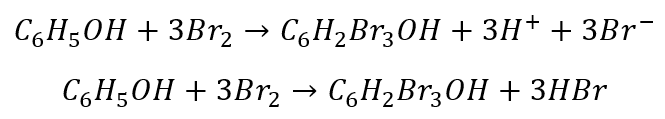
Taka praktyka jest często spotykana i w dalszej części spotkamy się z nią ponownie.
Ale najpierw pokażę, że dokonując takich operacji dokonujemy pewnego paradoksu. Zastanówmy się: skąd pochodzą elektrony w równaniu redukcji? „Wyciągamy” je z cząsteczki fenolu. Ale w równaniu utleniania pojawiają się aniony bromkowe – te, które wcześniej wygenerowaliśmy używając elektronów zabranych wcześniej od cząsteczki fenolu. Czyli wraz z tymi anionami bromkowymi do cząsteczki fenolu wracają wcześniej zabrane elektrony! Nie wszystkie co prawda, ale część zabranych „wkładamy” z powrotem. A ponieważ równania połówkowe to tylko matematyka, a nie rzeczywistość, to pomyślmy teraz razem. Pytanie: czy ma znaczenie czy ja najpierw zabiorę elektrony cząsteczce fenolu i przekażę je atomom bromu, a następnie użyję tych atomów bromu wzbogaconych w elektrony w równaniu utleniania i zwrócę część zabranych elektronów, czy też mogę w równaniu utleniania użyć obojętnych atomów bromu i po prostu zabrać mniej elektronów?
Czyli tak:
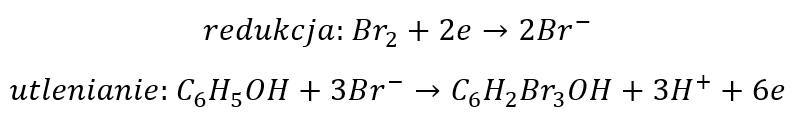
Czy tak:
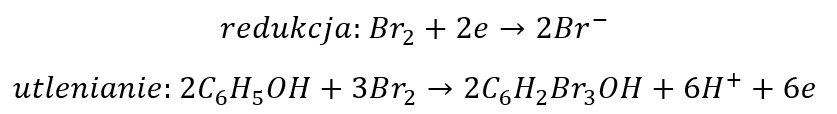
Czy to coś zmienia? Sprawdźmy. W pierwszym przypadku doszliśmy do takiego równania reakcji:
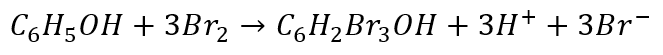
A co otrzymamy w 2 przypadku? Pomnóżmy i zsumujmy.
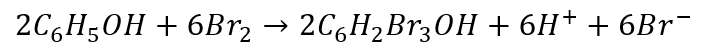
A w postaci najmniejszych liczb całkowitych:
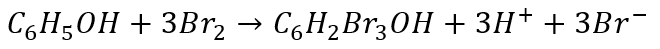
I to pomimo tego, że równania reakcji połówkowej utleniania się różnią, to dochodzimy do tego samego równania reakcji redoks. Dlaczego? Z tego samego powodu, o którym wspominałem wcześniej: równania połówkowe nie mówią nam nic o ścieżce reakcji. Służą tylko do bilansowania. To tylko matematyka - otrzymaliśmy równoważne układy równań połówkowych, więc muszą prowadzić do tego samego rozwiązania.
Taki zabieg z przenoszeniem anionów halogenkowych jest stosowany na przykład w reakcji haloformowej, jak w reakcji acetonu z jodem w środowisku zasadowym:
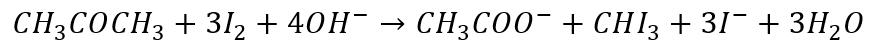
I równania połówkowe:
Redukcja:
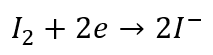
I utlenianie:
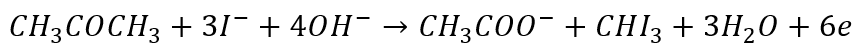
Jest to bardziej tradycyjna forma. Ale także tutaj Równania połówkowe to tylko matematyka. Możemy legitnie zapisać alternatywne równania reakcji połówkowych:
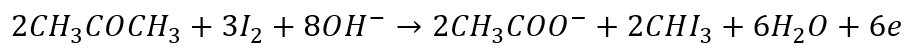
W reakcji jodoformowej utleniaczem wcale nie jest jod. A jednak zapisujemy go w równaniach połówkowych… Utleniaczem jest anion IO- powstający w wyniku dysproporcjonowania jodu w zasadowym środowisku:
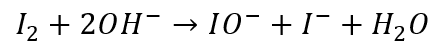
Gdy wrzucić jod do zasady to otrzymujemy bezbarwny roztwór. Właśnie wskutek reakcji dysproporcjonowania. Jeśli do tego roztworu dodać metyloketonu, to powstaje osad jodoformu, a przecież jodu, tradycyjnie zapisywanego jako substrat – w układzie nie było… A mechanizm reakcji jodoformowej jest znacznie bardziej skomplikowany… I jeśli prześledzimy, to w osobnym artykule poświęconym reakcji haloformowej, to sami zauważymy, że rzeczywista ścieżka reakcji nie ma nic wspólnego z tym, co zapisujemy w równaniach połówkowych…
Na koniec moja prośba do nauczycieli. Zarówno tych szkolnych, jak i akademickich: nie traktujmy równań reakcji połówkowych (elektrodowych) jak świętość. To jest tylko narzędzie do bilansowania. Nic za to nie mówi o ścieżce reakcji. Mówimy, że utleniamy alkohole dichromianem potasu w zakwaszonym środowisku. Ale jak się przyjrzymy bliżej, to wcale nie aniony dichromianowe działają utleniająco itd. Dichromianu potasu użyliśmy tylko by przygotować odczynnik utleniający. Bo każda reakcja ma jakiś swój mechanizm, a reakcje połówkowe (elektrodowe) o nim nic nie mówią. Nawet ta wymiana elektronów w wielu przypadkach fikcyjnym założeniem, ale o tym to napiszę już innym razem. I mam nadzieję, że niedługo. Jestem znowu w formie!
A! Jeszcze muszę wyjaśnić, dlaczego niektórzy nie potrafią zbilansować tej redukcji nitro benzenu. Ta trudność występuje tylko wtedy, gdy używać wsteczniczej metody za stopniami utlenienia. Niektórzy z góry błędnie zakładają, że stopień utlenienia azotu w nitrobenzenie jest równy V.

I dochodzą sprzeczności:
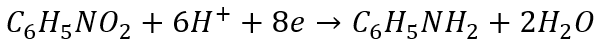
Bo przecież zgadzają się atomy, ale ładunek elektryczny nie zgadza się po obu stronach…A tymczasem w wyniku nitrowania azot obniżył swój stopień utlenienia z V na III…
I teraz dopiero się zgodzi:
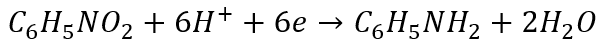
Ale problem mają tylko ci, którzy nie wiedzą, że to problem, którego nie ma, tylko problem stworzony przez samą edukację szkolną. Bo ci, którzy do bilansu jonowo-elektronowego podchodzą jak należy – tego problemu nie mają. Z bilansowania reakcji redoks w ogóle zrobiono potwora edukacyjnego: z prostej rzeczy zrobiono problem. A tak chyba być nie powinno. Po prostu pora skończyć z wstecznictwem i skupić się na tym, co ma być. Czyli na bilansie jonowo-elektronowym, w którym nie używa się stopniu utlenienia. A stworzenie potwora edukacyjnego polega na wsteczniczym stosowaniu stopni utlenienia i jednoczesnej próbie zastosowania ich do metody jonowo-elektronowej, która z natury ma ich nie używać. I tak metoda, która została wprowadzona po to, by ułatwić bilansowanie - w praktyce szkolnej skomplikowała prostą rzecz.
A przypominam, że stopnie utlenienia to jest pojęcie jeszcze bardziej fikcyjne, a przede wszystkim zazwyczaj niepotrzebne. Stopnie utlenienia nie istnieją. A skoro coś nie istnieje, to jaki jest sens nad nim gdybać: ile wynosi stopień utlenienia atomu X – no ile może wynosić coś, co nie istnieje? Przypisujemy je, ale ich nie ma, ponadto nawet obliczając wyimaginowane ładunki elektryczne - często są dyskusyjne, bo który atom powinien zabrałby elektrony? Przede wszystkim nie odnoszą się do rzeczywistości, a ponadto wynik zależy od skali elektroujemności. Prawie zawsze stosuje się Paulinga, której miejsce jest w lamusie, ale jak użyć innej skali, to atom, którego stopień utlenienia wyznaczamy na podstawie skali Paulinga może mieć zupełnie inny stopień utlenienia w tym samym związku, gdy użyć innej skali… Zobaczmy na definicję stopnia utlenienia: taki ładunek, jaki istniałby – no kurde! Jaki istniałby, a nie jaki jest! Ponadto, jak wspomniałem, często są trudne do ustalenia. Dlatego zrezygnowano ze stopni utlenienia przy bilansowaniu redoksów. Bo często są zawodne i nie zawsze pozwalają rozwiązać problem. A czy bilans reakcji w probówce zależy od stopni utlenienia? Jak bilans reakcji mógłby zależeć od czegoś, co nie istnieje? Więc nie zależy. Utrudniamy sobie (lub utrudniamy innym) życie czymś takim. Stosujemy metodę jonowo-elektronową, w której do zbilansowania równania reakcji redoks wystarczy wykonać bilans masy i bilans elektronowy. Albo raczej: stosujmy, bo z tym stosowaniem to jest w praktyce źle, a przeważnie: bardzo źle. A tak wiele osób nie jest w stanie ruszyć redoksa, jeśli najpierw nie ustali stopni utlenienia, albo nie jest w stanie ich ustalić. Bo nikt im powiedział, że w ogóle nie trzeba wiedzieć, co to jest stopień utlenienia, żeby zacząć bilansować reakcję redoks. Żeby określić, co się utlenia, a co redukuje. Tę kwestię wyjaśniam tutaj.
PS a jeszcze w kwestii stopni utlenienia. Można samemu się przekonać i policzyć elektrony: gdyby atom azotu miał V stopień utlenienia w nitrobenzenie, to nitrobenzen nie byłby związkiem, tylko kationem o ładunku 2+. A to oznacza, że występowałby tylko w solach, np. [C6H5NO2]SO4 albo [C6H5NO2]Cl2, a nie jako samodzielny związek.
PPS a jeszcze w kwestii mechanizmu reakcji jodoformowej – czy widzimy gdzieś etap, w którym byśmy wyciągali elektrony z cząsteczki acetonu i przekazywali je atomom jodu? Niby tak, ale przyjrzyjmy się – czy atom jodu rzeczywiście na tym zyskuje elektrony, a cząsteczka organiczna traci? Ale tę kwestię szerzej omówimy następnym razem.