
Mg(OH)2 - mocny czy słaby elektrolit?
Tym razem zajmujemy się tematem Mg(OH)2 – czy to mocny, czy słaby elektrolit?
Artykuł ten został opublikowany także w czasopismie „Chemia w szkole”.

Chociaż wiele osób oczekuje systemu zero-jedynkowego, a więc odpowiedzi: tak lub nie, to sprawa wymaga nieco dłuższej dyskusji oraz analizy tematu. Tym razem postanowiłem jednak podzielić artykuł na dwie części: w pierwszej skupić się na tym, co powinni wiedzieć uczniowie (i nauczycieli, by wiedzieli co przekazać), by czuć się usatysfakcjonowani, a nauczyciele by wiedzieli, ile przekazać, by ich usatysfakcjonować. A dalej będzie też coś dla osób zainteresowanych tematem trochę szerzej. Bo tak się składa, że najczęstsze pytanie jakie dostaję to „a na maturze to jak mamy pisać?” lub „a do matury to jak mamy ich uczyć?”. Cóż, nie wiem ile razy będę musiał pisać i pokazywać, że takie pytania jak i strach nie należą do grupy uzasadnionych, a przypominam, że dezinformację o zasadach przeprowadzania i oceniania egzaminu maturalnego niektórzy wprowadzają celowo, bo na tym zarabiają. Pokażę też z czym z poruszonych zagadnień, jak dotąd, zetknęliśmy się na maturze, bo często wiele rozterek wynika z chęci podjęcia tego egzaminu. Ale do rzeczy.
Jeśli interesuje Cię tylko odpowiedź tak lub nie, to przejdź do punktu 2.
Jeśli interesuje Cię temat, to zacznij od punktu 1.
1. Wersja dla zainteresowanych tematem
1.1. Związki jonowe w roztworach. Czym właściwie jest dysocjacja?
Związki jonowe, takie jak NaCl, są w wodnych roztworach zdysocjowane na jony. Związki jonowe nie tworzą cząsteczek, tylko jak nazwa wskazuje, są zbudowane z jonów, np. Na+ i Cl- w przypadku NaCl, a nie z cząsteczek Na-Cl. W wodnym roztworze NaCl także mamy do czynienia z jonami Na+ i Cl-. Wiele osób, nie tylko wśród uczniów, twierdzi, że NaCl wsypany do wody zdysocjuje całkowicie, bo jest to mocny elektrolit. Ze szkoły bardzo często wynosimy błąd: mylenie rozpuszczania z dysocjacją. I o tym można naprawdę dużo napisać. Nic dziwnego, bo w podręcznikach szkolnych w także na Wikipedii znajdziemy takie rysunki (Sorry, Adam ;-)).
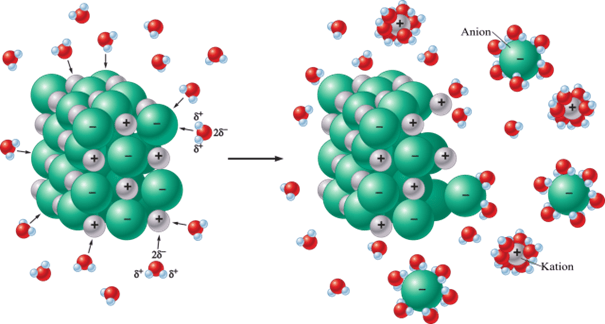
Rysunek ten nie jest sam z siebie zły – on bywa nie do końca odpowiednio rozumiany i jest to jedna z przyczyn mylenia rozpuszczania z dysocjacją elektrolityczną. Temat mylenia pojęć i wynikających z niego błędnych przekonań poruszyłem już w artykule „Czy Ca(OH)2 dysocjuje?”.
Wsypując łyżeczkę soli do szklanki z wodą stwierdzamy, że NaCl rozpuszcza się w wodzie, czy że dysocjuje w wodzie? A jeśli dysocjuje, to czy ulega dysocjacji całkowicie?
To co obserwujemy to jest rozpuszczanie NaCl. A nie dysocjacja. Czy ktokolwiek słyszał w domu: "Janek, zdysocjuj mi tam łyżkę soli w tej zalewie do ogórków. Czubatą!".
O dysocjacji mówimy w roztworze, a że NaCl ma ograniczoną rozpuszczalność w wodzie, to gdy wprowadzimy za dużą ilość NaCl w stosunku do rozpuszczalnika, to tylko część NaCl przejdzie do roztworu (rozpuści się). Jeśli do kilograma wody wsypać 1 kg soli kuchennej, to w temperaturze pokojowej rozpuści się ok. 1/3 wsypanej ilości. A 2/3 pozostanie na dnie. Ale to nie znaczy że stopień dysocjacji wynosi ok. 1/3, bo tylko 1/3 się rozpuściła („zdysocjowała”). Bo o stopniu dysocjacji mówimy w roztworze. Czyli nie interesuje nas, ile się rozpuściło, tylko jak zachowuje się to, co się rozpuściło. Dlatego jest ważne by odróżniać pojęcia rozpuszczanie i dysocjacja. Zauważmy, że gdyby chodziło o cukier, to nikt by nie mówił o niczym innym niz rozpuszczaniu. I chociaż na samym początku nauki o roztworach mówiono nam o rozpuszczaniu soli w wodzie, to potem nasze myślenie jest często skrzywiane przez wprowadzenie pojęcia dysocjacji.
NaCl rozpuszcza się w wodzie i jest to coś, czego na podstawie obserwacji jesteśmy pewni, ale czy dysocjuje w wodzie to inna sprawa. Niezależna od rozpuszczania. Taki KClO4 w wodzie się słabo rozpuszcza, ale jest mocnym elektrolitem, ze względu na to jak zachowuje się w roztworach. W roztworach wodnych NaCl ulega dysocjacji, stąd są w nim jony Na+ i Cl-. Odbiegłem trochę od tematu, ale mam ku temu powód. NaCl jest związkiem jonowym, a skoro nie ma cząsteczek NaCl, to w roztworze wodnym też ich nie ma. Cóż miałoby przeszkadzać temu, by stopień dysocjacji takiego NaCl w wodzie wynosił 100%? Przecież jako związek jonowy jest „zdysocjowany z natury”, czyli zbudowany z jonów, które uwalniają się z sieci krystalicznej i przechodzą do roztworu. Możemy to tak rozpatrywać, jak widać na rysunku Adama, który sam z siebie jest dobry, ale nie do końca dobrze rozumiany przez gawiedź szkolną - i niestety, także przez część nauczycieli. Rutyna bywa zła.
Dysocjują w wodzie nie tylko związki jonowe w niej rozpuszczone, ale także liczne związki kowalencyjne, jak np. chlorowodór HCl. Takie związki także mogą prawie całkowicie dysocjować – z tego powodu kwas solny zaliczamy do kwasów mocnych.
Ale mamy także takie związki jak H2SO4. W wodnych roztworach cząsteczki H2SO4 dysocjują przekazując swój proton cząsteczce wody.
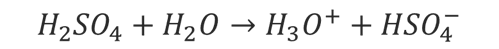
Ok, ale H2SO4 to związek kowalencyjny, gdzie każdy etap ma swoją stałą dysocjacji. Duże wartości stałej dysocjacji trudniej wyznaczyć ze względu na metody wyznaczania stałych dysocjacji, dlatego dane literaturowe co do wartości Ka1 są rozbieżne i wiele tablic w takich przypadkach nie podaje wartości Ka1 dla H2SO4. Pierwsza stała dysocjacji i tak ma małe znaczenie praktyczne, bo czy na podstawie użytej wartości stałej kwas jest obliczony stopień dysocjacji wynosi 99%, czy 99,5%, to i tak stężenie H3O+ w roztworze jest praktycznie identyczne: 0,099 czy 0,0995 to nadal praktycznie 0,1 mol/dm3. Czyli kwas zachowuje się tak samo. Łatwiej wyznaczyć mniejsze stałe dysocjacji. Druga stała dysocjacji dla H2SO4 wynosi 0,01 w 25 °C – jon HSO4- jest kwasem średniej mocy i nie zmienia to faktu, że H2SO4 to kwas mocny w wodnych roztworach.
Ale tak jak mamy kwasy wieloprotonowe, tak samo mamy wodorotlenki, w których na jeden kation metalu przypadają co najmniej dwa jony wodorotlenkowe, czyli OH-. Przykładami są Ca(OH)2, Mg(OH)2 czy Sr(OH)2. Związki jonowe, jak często słyszymy, i wiele oburza się na hasło o etapowej dysocjacji wodorotlenków. „Bo to związki jonowe!”. Ponadto patrząc na rysunek Adama zaczerpnięty z Wikipedii i jego odpowiedniki w podręcznikach, to takie oburzenie wydaje się uzasadnione. Dlatego też zwykle widzimy w podręcznikach takie równania dysocjacji:
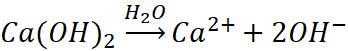
Możemy sobie wyobrazić rozpuszczanie takiego Ca(OH)2 jako przechodzenie jonów Ca2+
i OH- z sieci krystalicznej do roztworu. Na pewnym etapie edukacji nie lubię o pewnych rzeczach mówić ludziom, ale też nie mówię im nieprawdy. Jednakże jedni chcą po prostu wiedzieć: tak lub nie, a inni jeszcze zrozumieć. A tutaj skupiamy się na tych, którzy chcą zrozumieć.
Zastanówmy się, czy widzieliśmy kiedyś stałą dysocjacji np. NaCl? Stałe dysocjacji, z którymi większość z nas się spotyka dotyczą ogólnie połączeń kowalencyjnych: cząsteczek lub jonów jak np. HSO4-. Czy można więc mówić o stopniu dysocjacji NaCl, albo innego jonowego związku w roztworze, skoro nie ma cząsteczek Na-Cl, tylko po prostu jony przechodzące z sieci krystalicznej do roztworu?
Odpowiemy zaiste, że nie, bo skoro mamy sieć zbudowaną z jonów, to nic nie przeszkadza w tym, by jony bezpośrednio przechodziły do roztworu. Co innego np. w przypadku kwasu cytrynowego, który jest związkiem kowalencyjnym – do roztworu wodnego przechodzą cząsteczki, które dopiero w roztworze mogą przechodzić w jony. Zatem w przypadku wodorotlenków też tak powinno być jak w przypadku NaCl.
Jednakże czym jest dysocjacja elektrolityczna? To nie jest sam rozpad na jony, choć często jest z tym utożsamiana, choć nie zawsze właściwie. O dysocjacji mówimy dopiero wtedy, kiedy jony mogą się poruszać niezależnie od siebie. Wtedy, kiedy kation i anion poruszają się każdy „swoją drogą”. Weźmy pod uwagę, że kation i anion to cząstki przeciwnych ładunkach elektrycznych, czyli takie, które się przyciągają elektrostatycznie (siłą Coulomba). A zatem tak jak jony przyciągają się wzajemnie w sieci krystalicznej, tak samo drobiny o przeciwnych ładunkach muszą się przyciągać w roztworze – przecież siła kulombowska nie znika w magiczny sposób w roztworze – po przejściu do roztworu nadal działają prawa fizyki. Im bliżej siebie znajdują się jony przeciwnych znaków, tym silniej się przyciągają. Przyciągający się kation i anion mogą więc się spotkać i pozostać razem…
Jony przeciwnych znaków mogą utworzyć zespół zwany parą jonową. Na rysunku poniżej przedstawiłem różnicę pomiędzy samym istnieniem jonów a dysocjacją elektrolityczną.
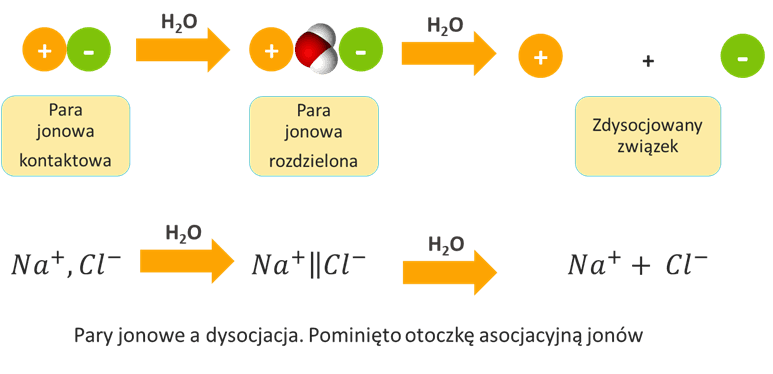
Par jonowych nie należy mylić z cząsteczkami! To nie to samo. Para zbudowana z jonów Na+ i Cl- to nie jest cząsteczka chlorku sodu (Na–Cl). Pary jonowe, czyli takie nibycząsteczki NaCl obecne są np. w parach NaCl. W roztworach to cząsteczki rozpuszczalnika mogą powodować rozsuwanie jonów na taką odległość, że mogą się one zacząć poruszać osobno, bo siłą przyciągania jest za słaba. I dopiero to nazywamy dysocjacją – nie sam fakt tworzenia jonów, tylko ich rozdzielenie w taki sposób, by mogły się poruszać niezależnie od jonu przeciwnego znaku. Woda jest rozpuszczalnikiem o dużej polarności i do tego o bardzo małych cząsteczkach, jednymi z najmniejszych w ogóle cząsteczek, a zatem o takich cząsteczkach, które „wszędzie się wcisną”. Z tego powodu woda wyjątkowo sprzyja dysocjacji elektrolitycznej, bo bardzo skutecznie rozdziela jony od siebie.
A jednak nawet w roztworach wodnych mogą istnieć pary jonowe, a to oznacza, że nawet związki jonowe mogą nie być w pełni zdysocjowane w wodzie. Za przewodnictwo elektryczne w roztworach odpowiadają wolne jony, bo to one są nośnikami ładunku elektrycznego. Zauważmy, że jony będą się poruszać w kierunku odpowiedniej elektrody w polu elektrycznym. A para jonowa? Para jonowa np. Na+, Cl- nie ma ładunku elektrycznego. Jest elektrycznie obojętnym tworem – nie porusza się w polu elektrycznym, nie jest nośnikiem ładunku, jest takim „jonem obojnaczym”. Mierząc przewodnictwo roztworu można określić stężenie par jonowych w roztworze. W niektórych przypadkach może ich być dużo, np. stwierdzono, że w nasyconym roztworze CaSO4 aż 33% jonów tworzy pary jonowe, co oznacza, że stopień dysocjacji CaSO4 w wodzie (w nasyconym roztworze zwanym wodą gipsową) wynosi 67%. Tak, taki CaSO4 posiada swoją stałą dysocjacji, która opisuje równowagę pomiędzy parami jonowymi, a wolnymi jonami. Wobec faktu istnienia par jonowych nasz rysunek z rozpuszczaniem kryształu jonowego możemy wzbogacić o fakt, że przecież kryształ mogą opuszczać nie tylko pojedyncze jony, ale także pary jonowe – nie muszą przecież one powstawać dopiero w roztworze, gdy jony spotkają się ze sobą, lub znajdą odpowiednio blisko. Pary jonowe występują także w roztworach związków kowalencyjnych, zdolnych do dysocjacji elektrolitycznej. Dla przejrzystości opisu w dalszej części nie będziemy wyróżniać par jonowych kontaktowych i rozdzielonych, tylko po prostu pary jonowe.
1.3. Dysocjacja wodorotlenków
Teraz przejdźmy do wodorotlenków, rozpatrzmy wodorotlenek wapnia na początek. Czy wobec przyciągania się jonów możemy mówić np. o tym, że Ca(OH)2 może dysocjować etapami, podobnie jak w przypadku H2SO4? Jak już mówiłem: wielu dydaktyków oburzy się na stwierdzenie, że tak.
Jony Ca2+ także wzajemnie się przyciągają z jonami OH-, zarówno w roztworze, jak i poza nim. Możemy więc sobie wyobrazić tworzenie pary jonowej pomiędzy jonem Ca2+ a OH-. A skoro tak, to możemy mówić o etapach dysocjacji wodorotlenku!
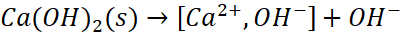
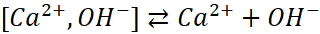
Celowo zamiast Ca(OH)+ albo [Ca(OH)]+ napisałem [Ca2+,OH-], żeby czytelnik nie odebrał tego jako połączenie kowalencyjne, tylko para jonów Ca2+ i OH- o sumarycznym ładunku +1. Oczywiście możemy to zapisać tak, jak bardziej jesteśmy przyzwyczajeni:![]()
![]()
Ale po prostu chciałem wskazać na jonowy charakter tego połączenia. Wynika z tego, że pomimo jonowego charakteru, taki wodorotlenek wapnia powinien posiadać… stałe dysocjacji. Oczywiście w szkolnych tablicach ich raczej nie znajdziemy. Ale znajdziemy w innych tablicach. Pierwszy etap jest zasadzie nieodwracalny, więc zwykle nie określa się pierwszej stałej dysocjacji, podobnie jak to ma miejsce w przypadku H2SO4. Zobaczmy, co o wodorotlenkach dowiadujemy z tablic:
Związek | Kd1 | Kd2 |
Be(OH)2 | b.d. | 5,0∙10-11 |
Mg(OH)2 | nie określa się | 2,5∙10-3 |
Ca(OH)2 | nie określa się | 4,3∙10-2 |
Sr(OH)2 | nie określa się | 0,16 |
Ba(OH)2 | nie określa się | 0,25 |
A pewne szkolne źródło podaje takie oto cuda…

Nie muszę chyba tłumaczyć, że ktoś tutaj błędnie posługiwał się tablicami przy sporządzaniu tej tabelki.
A co z wodorotlenkami litowców? Pomijając LiOH wodorotlenki te praktycznie nie mają skłonności do tworzenia par jonowych w wodnych roztworach i mają bardzo duże wartości stałych dysocjacji, ale do ich tematu jeszcze wrócę w osobnym artykule. Przyjrzyjmy się wartościom drugiej stałej dysocjacji – jej wartość rośnie ze wzrostem liczby atomowej metalu – jest to przecież zgodne z tym czego się uczymy: rośnie moc tych wodorotlenków jako elektrolitów. Najmniejszą skłonność do tworzenia par jonowych mają jony Ba2+ z jonami OH-, a to oznacza, że para utworzona przez te dwa jony najłatwiej się rozpada, czyli w największym stopniu dysocjuje. Pierwszy etap dysocjacji tych wodorotlenków możemy potraktować jako zachodzący całkowicie. Czy tę tendencję do wzrostu skłonności par jonowych do dysocjacji można jakoś wyjaśnić? Tak. Oddziaływanie kulombowskie jest skuteczniejsze gdy oddziałujące ładunki elektryczne są skupione w jakimś punkcie. Tymczasem nasze kationy mają jakąś wielkość, jakiś rozmiar i zawsze taki sam ładunek: +2. Skuteczniej oddziaływuje mniejszy kation Mg2+ niż większy Ca2+, że o Ba2+ już nie wspomnę. Ładunek jonu Ba2+ jest najbardziej rozmyty ze względu na największy rozmiar tego kationu. Oddziałuje on więc z jonami OH- najsłabiej z wymienionych. Uzupełniająco dodam, że Mg(OH)2 wykazuje pewne cechy kowalencyjności.
Im niższe stężenie roztworu, tym bardziej coś zdysocjowane. Jak możemy sami policzyć, taki wodorotlenek magnezu jest w roztworze wodnym niemal całkowicie zdysocjowany na jony Mg2+ - stopień dysocjacji pary Mg(OH)+ przekracza 92%, co oznacza, że stężenie jonów OH- jest tylko o 4% niższe, niż obliczone z tradycyjnie spotykanego w książkach równania dysocjacji:
![]()
Musimy jednak pamiętać, że tak wysoki stopień dysocjacji wynika z małego stężenia nasyconego roztworu Mg(OH)2 – w 25 °C wynosi ono zaledwie 0,00011 mol/dm3. Przy tym samym stężeniu stopień dysocjacji [Ca2+,OH-] przekracza 99%. Żeby być miarodajnym musimy porównać dysocjację par jonowych przy tym samym stężeniu. W tabeli poniżej zestawiłem wyniki obliczeń stopnia dysocjacji odpowiednich par przy stężeniu wyjściowego wodorotlenku 0,01 mol/dm3. Dane dotyczą 25 °C.
Para jonowa | Stopień dysocjacji | pH roztworu (25 C°) |
Mg(OH)+ | 18%* | 12,07* |
Ca(OH)+ | 71% | 12,23 |
Sr(OH)+ | 89% | 12,28 |
Ba(OH)+ | 93% | 12,29 |
*W przypadku Mg(OH)2 jest to wartość hipotetyczna – gdyby związek był w stanie osiągnąć takie stężenie. Stężenie nasyconego roztworu Mg(OH)2 jest znacznie niższe od 0,01 mol/dm3.
Gdyby wszystkie wodorotlenki dysocjowały tak, jak jesteśmy uczeni, to roztwory wszystkich wodorotlenków miałyby pH zbliżone do 12,30. – nie byłby całkiem identyczne, ale bardziej zbliżone do 12,30. Różnice wynikałyby z jeszcze innego powodu, którego nie będę tutaj omawiał, ale zainteresowanych odsyłam do II części „Podstaw obliczeń chemicznych” mojego autorstwa.
Z powodu obecności par jonowych pH wody wapiennej (nasyconego roztworu Ca(OH)2 w wodzie) jest nieco niższe niż obliczone z rozpuszczalności albo iloczynu rozpuszczalności. Być może sami już mieliśmy okazję przekonać się o tym – dla zainteresowanych coś na końcu.
1.4. A co z hydrolizą w roztworach soli Mg2+ itp.?
W przypadku amoniaku nie mamy problemu, by jego stałą dysocjacji zasadowej (Kb) przeliczyć na stałą dysocjacji kwasowej (Ka) kationu amonowego, czyli stałą hydrolizy. Wystarczy skorzystać z zależności:![]()
Możecie mi nie wierzyć, ale kto nie wierzy niech sobie sam wyprowadzi, że analogiczna zależność pracuje dla sprzężonych par w omawianym przypadku. I tak jeśli rozpatrujemy:
![]()
To dla reakcji:
![]()
Obliczymy stałą dla takiej dysocjacji (czyli hydrolizy) następująco:![]()
Czyli w 25 °C: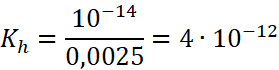
Jest to bardzo mała wartość, co oznacza, że i hydroliza musi zachodzić w bardzo małym stopniu. Jeśli wykonamy to samo dla anionu SO42-, to obliczymy jego stałą hydrolizy (dysocjacji zasadowej) i otrzymamy:
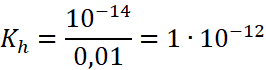
Wartość bardzo mała, ale co ważniejsze: zbliżona do stałej hydrolizy kationu Mg2+. Czy ktokolwiek mówi o hydrolizie w roztworze K2SO4? Właściwie nie, bo ze względu na bardzo małą stałą hydrolizy zachodzi ona w tak małym stopniu, że można ją zaniedbać, bo prawie nie zachodzi – roztwór K2SO4 ma odczyn praktycznie obojętny (pH nieznacznie ponad 7 i nie wykryjemy zasadowego odczynu np. fenoloftaleiną i wieloma innymi wskaźnikami). Z kolei w przypadku roztworów np. Mg(ClO4)2 pH będzie nieznacznie poniżej 7. Zarówno w przypadku Mg2+ jak i SO42- hydroliza jest pomijalna i jej skutków praktycznie nie obserwujemy. Dlatego choć niektórzy spodziewają się kwasowego odczynu soli Mg2+, takich jak MgSO4 („bo to sól mocnego kwasu, anion SO42- nie hydrolizuje”) to czeka ich zawód, bo jest prawie obojętny i ledwo można stwierdzić, że nie jest obojętny. Pospolitymi wskaźnikami nie odróżnimy wodnego roztworu Mg(NO3)2 od czystej wody.
To co do tej pory mówiliśmy, to i tak wersja uproszczona dla związków magnezu. W roztworach wodnych w większości przypadków nie ma prostych kationów metali, tylko są tzw. akwakompleksy. To one nadają kolor roztworom związków metali bloków d i f. Rzadko uwzględniamy akwakompleksy w równaniach reakcji, albo nawet ich występowanie w roztworze – często słyszymy o kationach metali. Jest to jednak duże uproszczenie. Zazwyczaj dysocjację np. MgSO4 widzimy zapisaną w taki sposób:![]()
![]()
Wiele soli ma budowę kowalencyjną – ich dysocjacja, którą zwykle zapisujemy w uproszczony sposób, nie polega tworzenie prostego kationu, albo kompleksowego kationu, tylko na wymianie ligandów: np. anionu chlorkowego na cząsteczkę wody. Np. chlorek miedzi jest związkiem kowalencyjnym, rozpatrzmy co się z nim dzieje po rozpuszczeniu w wodzie:
![]()
Pierwszy obojętny akwakompleks ulega dysocjacji z „odszczepieniem” anionu chlorkowego, co w rzeczywistości jest reakcją wymiany ligandów:![]()
Czyli w uproszczeniu:![]()
I drugi etap dysocjacji:
![]()
Czyli w uproszczeniu:
![]()
Chlorek miedzi(II) to bardzo wygodny przykład by pokazać pewne rzeczy niedowiarkom – ale zainteresowanych odsyłam do odpowiednich artykułów tutaj lub tutaj.
Wróćmy do naszego Mg(OH)2. Pierwszy etap dysocjacji możemy zapisać na dwa sposoby. Pierwszy:![]()
Gdy rozpatrywać połączenie pomiędzy Mg a OH jako kowalencyjne. A drugi, gdy rozpatrywać utworzenie pary jonowej:![]()
I drugi etap dysocjacji jako wymiana ligandów:![]()
Lub jako dysocjacja w prawdziwym znaczeniu:![]()
W roztworze Mg(OH)2 takie procesy zachodzą równolegle i mamy więcej indywiduów chemicznych, które można opisać w postaci uproszczonej jako Mg(OH)+ i zapisać powyższe równowagi jednym prostszym równaniem, bez względu na to, czy rozpatrujemy parę jonową czy też odpowiedni kompleks:
![]()
Proces, w którym w pierwszym etapie dysocjacji powstaje para jonowa [Mg(H2O)62+,OH-] prawdopodobnie dominuje nad procesem, w którym powstaje jon kompleksowy [Mg(H2O)5OH]+, stąd możemy to rozpatrywać jako zjawiska, w których biorą udział pary jonowe, jak było w opisie w poprzedniej części artykułu. Im bardziej kowalencyjny charakter wodorotlenku, tym stosunek par jonowych do jonów kompleksowych jest niższy.
Wracając do hydrolizy. Równowaga hydrolizy w roztworach soli magnezu opisaną wcześniej równaniem:![]()
Jest sumarycznym uproszczonym zapisem równowag:![]()
Oraz:![]()
Pewna liczba kationów [Mg(H2O)6]2+ tworzy z jonami OH- pary jonowe:![]()
Wiązanie anionów OH- w parę jonową powoduje przesunięcie równowagi dysocjacji wody w prawo – w układzie mamy więc nadmiar wolnych jonów H3O+ nad wolnymi jonami OH-, czyli odczyn kwasowy. Możemy to opisać jednym równaniem jako:![]()
Podkreślam jednak, że obie te reakcje zachodzą w bardzo małym stopniu. Do tego małym, że roztwory soli Mg2+ mają odczyn prawie obojętny.
Dopiero w przypadku np. soli Al3+, pochodzących od Al(OH)3, hydrolizę możemy rozpatrywać następująco:![]()
Porównajmy 4·10-12 z 1,4·10-5. To oznacza, że jon [Al(H2O)6]3+ jest w podobnym stopniu kwasowy, jak CH3COOH w wodzie! Stała dysocjacji kwasu octowego w wodzie wynosi 1,75·10-5. W przeciwieństwie do soli Mg2+, hydroliza w roztworach Al3+ jest zdecydowanie zauważalna, co przejawia się w odczynie roztworów Al3+ - znacznie odbiegających od obojętnego. Oznacza to, że rozcieńczony roztwór CH3COOH i rozcieńczony roztwór Al(NO3)3 o tych samych stężeniach i temperaturze – mają niemal równe pH.

Więcej znajdziesz na stronie: https://biomist.pl
Zwróćmy uwagę na różnicę zabarwienia w probówce z r-rem MgSO4 i w probówce z KAl(SO4)2. W ostatniej probówce (po prawej) widzimy zachowanie uniwersalnego papierka wskaźnikowego wobec roztworu MgSO4. Zielonkawożółty kolor świadczy o odczynie praktycznie obojętnym. Doświadczenie zostało starannie przygotowane (szczegóły na końcu artykułu). Ze względu na znacznie bardziej kowalencyjny charakter tego związku, jony kompleksowe zdecydowanie dominują parami jonowymi tworzonymi przez Al(H2O)6]3+ z jonami OH- i możemy hydrolizę rozpatrywać tak jak w równaniu powyżej.
A co w przypadku soli Ca2+, czy Ba2+? Jeśli obliczymy stałą hydrolizy dla odpowiednich reakcji:![]()
![]()
W roztworach soli wapnia czy baru zjawiska hydrolizy są jeszcze bardziej pomijalne niż w roztworach soli Mg2+ czy SO42-, czyli ta hydroliza (praktycznie) nie zachodzi. Odczyn takich roztworów jest praktycznie obojętny – jeszcze bardziej obojętny, niż w roztworze K2SO4(!) czy Mg(ClO4)2.
1.5. Co było na maturze?
Jak do tej pory nie pytano bezpośrednio o hydrolizę w roztworach soli magnezu, bo i nie ma specjalnej potrzeby. Ale z poruszonych zagadnień warto wspomnieć o zadaniu z czerwca 2016 roku:

I jeszcze bardziej warto zwrócić uwagę na zadanie z czerwca 2022 roku:


A więc rozpatrujemy tutaj hydrolizę w roztworach soli magnezu. Świat nie jest zero-jedynkowy i także maturzysta musi być tego świadom. Czy umiemy odpowiedzieć na to pytanie? Można dobrze odpowiedzieć i bez znajomości akwakompleksów, bowiem nawet do matury musimy wiedzieć, jak zmienia się charakter chemiczny wodorotlenków w okresach i grupach układu okresowego pierwiastków. Al(OH)3 ma bardziej kwasowy charakter niż Mg(OH)2 – a zatem jest przy okazji jest słabszym elektrolitem i hydroliza w roztworach Al3+ zachodzi w znacznie większym stopniu (i przy łatwo wykrywalnym). Czyli chętniej tworzy się hydroksokompleks [Al(H2O)5OH]2+ niż analogiczne kompleks magnezu. W uproszczeniu podane równanie hydrolizy zapiszemy jako:![]()
1.6. Jeszcze o parach jonowych, roztworach substancji kowalencyjnych i iloczynie rozpuszczalności
W roztworach związków kowalencyjnych, także mogą istnieć pary jonowe. Zerknijmy na tzw. schemat Winsteina na przykładzie dysocjacji HCl w wodzie:
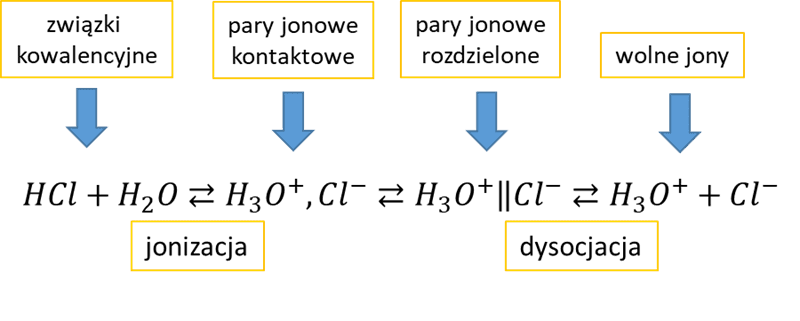
Rysunek ten należy traktować poglądowo. Zapisy dysocjacji, których uczymy się w szkole, np.![]()
są uproszczeniem problemu, które zresztą pracuje dobrze i nie zamierzam z tym walczyć, jakby niektórzy po mnie oczekiwali – chcę tylko zwiększyć świadomość. Czym różni się para jonowa kontaktowa od rozdzielonej? Jeśli powstałe jony kontaktują się bezpośrednio ze sobą, to nazywamy je parą kontaktową. Cząsteczki rozpuszczalnika mogą jednak „wcisnąć się” pomiędzy nie, np. załóżmy, że pomiędzy jony H3O+ i Cl- wciśnie się cząsteczka wody:
H3O+,H2O,Cl-
Ale siła Coulomba nadal jest na tyle duża, że jony tworzą parę wraz z tą cząsteczką wody i wraz z tą cząsteczką wody pomiędzy poruszają się tworząc takie trio. Ale nadal jest to para jonów. Istnieje pewna krytyczna odległość, powyżej której siła przyciągania jest już na tyle słaba, że jony poruszają się niezależnie od siebie. To tak jak z dwoma magnesami sczepionymi ze sobą – na jaką odległość je można rozsunąć, aby siła grawitacji była większa niż siła ich przyciągania i magnes dolny odpadł od górnego. Możemy wsuwać np. kartki papieru pomiędzy nie i policzyć, ile kartek potrzeba, by jeden odpadł od drugiego. Odległość ta różna dla różnych ośrodków, które umieścimy pomiędzy magnesami. Tak samo z jonami w roztworach – w zależności od przenikalności pola elektrycznego w danym rozpuszczalniku odległość na jaką cząsteczki rozpuszczalnika mogą rozdzielić jony nie powodując, że zaczynają się poruszać niezależnie od siebie – jest różna, czyli mniej lub więcej cząsteczek rozpuszczalnika musi znaleźć się pomiędzy kationem i anionem, aby przestały stanowić parę poruszającą się razem. Dla wody jako rozpuszczalnika ta odległość jest niewielka. Tak więc zależy to zarówno od natury substancji rozpuszczonej, jak i natury rozpuszczalnika. Dodajmy, że także w zależności od natury substancji rozpuszczonej i rozpuszczalnika określonego typu par jonowych w roztworze może nie być wcale, lub określonego typu par jonowych może nie być.
Powszechnie wynosimy ze szkoły, czy nawet studiów, że iloczyn rozpuszczalności możemy przeliczyć na rozpuszczalność. Ale gdyby było tak łatwo, to byłoby zbyt piękne, aby prawdziwe. Iloczyn rozpuszczalności rozpatruje równowagę pomiędzy osadem, a jonami. Nie uwzględnia się przy tym, że w roztworze mogą być (i często są) pary jonowe – rzeczywista rozpuszczalność może być znacznie wyższa, niż obliczona z iloczynu rozpuszczalności. Jeśli lubimy się bawić, to możemy sami poszukać odpowiednich danych i przeliczyć rozpuszczalność na iloczyn rozpuszczalności lub na odwrót dla różnych substancji i porównać wyniki z tablicami. Różnice da się zauważyć prawda? Zainteresowanych tematem odsyłam do rozdziału 5.15 w uzupełnieniu w II części „Podstaw obliczeń chemicznych”.
2. Wersja dla niecierpliwych
Wiele osób zadaje sobie pytanie: czy Mg(OH)2 to mocny elektrolit/mocna zasada? Pytanie to nie jest bezzasadne, bo wg jednych źródeł Mg(OH)2 to mocny elektrolit, wg innych nie. W tym artykule rozkminiliśmy to.
Zacznijmy od pojęcia zasada. Nawet licealista (a więc i maturzysta), musi wiedzieć, że pojęcie to ma dwa znaczenia:
1) tak nazywamy przeciwieństwo kwasu w teoriach kwasowo-zasadowych,
2) tak nazywamy roztwór wodny dobrze rozpuszczalnego wodorotlenku.
Inaczej mówiąc zasada sodowa i wodorotlenek sodu to nie to samo. Wodorotlenek sodu to w temperaturze pokojowej ciało stałe o wzorze NaOH, a zasada sodowa to wodny roztwór tego związku NaOH(aq). Ze względu na stężenie roztworu zasadę sodową dzielimy na rozcieńczoną i stężoną zasadę sodową.
W tym drugim znaczeniu coś jest zasadą albo nie, a wodorotlenek magnezu jako trudno rozpuszczalny w wodzie nie tworzy zasady. Ale z drugiej strony - pytanie, a gdzie jest granica? Bo np. fenoloftaleina w mleku magnezjowym (zawiesinie Mg(OH)2 w wodzie) przyjmuje całkowicie malinowe zabarwienie (pH>10). Możnaby wiec przyjąć, że jeśli w jakimś roztworze wodorotlenku taka fenoloftaleina tak sobie, albo jako tako się barwi, to taki roztwór to nie jest zasada.
A jeśli mówimy o zasadzie wg pierwszego znaczenia, to we współczesnych teoriach kwasowo-zasadowych (a więc w obowiązującej teorii Broensteda-Lowry’ego) to nie wodorotlenki są zasadami, tylko jony OH-. Jon OH- jest zawsze zasadą tej samej mocy, a do tego zasadą mocną i nie ma znaczenia skąd pochodzi. Tylko w znaczeniu pierwszym mówimy o mocy zasad, a w znaczeniu drugim: coś albo jest zasadą, albo nią nie jest. Tylko w teorii Arrheniusa, która jest teorią historyczną i szybko zastąpioną przez inne teorie, Mg(OH)2 zaliczamy do zasad.
Niezależnie od tego, czy w teoriach kwasowo Mg(OH)2 zaliczymy do zasad lub nie, to i tak możemy mówić o mocy tego związku na podstawie skłonności do dysocjacji. Ponieważ w roztworze wodnym dysocjuje on praktycznie całkowicie, to jest to elektrolit mocny. W roztworach soli magnezowych w wodzie takich jak MgCl2 czy MgSO4 obserwuje się praktycznie obojętny odczyn. W skrócie: wodorotlenek magnezu jest elektrolitem mocnym, ale słabo w wodzie rozpuszczalnym – moc elektrolitu określamy na podstawie zachowania w roztworze, a nie na podstawie tego, ile się go rozpuści. Jeszcze raz uczulam: nie myl rozpuszczania z dysocjają!
Wodorotlenek magnezu jest mocnym elektrolitem mocnym, ale słabo rozpuszczalnym. Wodne roztwory soli Mg(OH)2 z mocnymi kwasami mają odczyn praktycznie obojętny. Hydroliza kationowa w takich roztworach zachodzi w pomijalnie małym stopniu.
Jeśli interesuje Cię, co poruszono do tej pory na maturze z tematu Mg(OH)2 i hydrolizy soli magnezowych – wróć do punktu 1.5.
Opis doświadczenia: wszystkie probówki i kolby miarowe wytrawiono rozcieńczonym HNO3, następnie wypłukano wodą destylowaną a następnie wodą demineralizowaną o przewodnictwie 0,066·10-3 µS/cm. Probówki i kolby wysuszono w temperaturze 130 °C w atmosferze powietrza. Wodę demineralizowaną odpowietrzono przez przedmuchiwanie argonem przez 30 min. Kolby przedmuchiwano argonem przez 10 min i przygotowano roztwory o stężeniu 0,2 mol/dm3 używając MgSO4·7H2O czda i KAl(SO4)2·12H2O czda w atmosferze argonu. Kolby użytej do sporządzenia r-r CH3COOH nie przedmuchiwano argonem. 4 Probówki wypełniono argonem i umieszczono po 10 cm3 odpowiedniej cieczy: wody demineralizowanej (odpowietrzonej) (probówka 1), roztworu MgSO4 (probówki 2 i 5), roztworu KAl(SO4)2 (probówka 3). Roztwór kwasu octowego umieszczono w probówce wypełnionej powietrzem. Wodę demineralizowaną i roztwory soli pobierano za pomocą strzykawki z igłą w atmosferze argonu.